Epilepsia este o afecțiune neurologică cronică caracterizată prin crize de alterare a stării de conştienţă, asociind manifestări motorii, sensitive sau senzoriale şi datorate unor descărcări paroxistice şi sincrone ale unor populaţii neuronale mai mult sau mai puţin extinse.
Problemele pe care le poate prezenta un bolnav cu epilepsie sunt: convulsii, insomnii, halucinaţii, tahicardie, confuzie, dispnee.

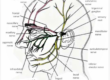
Sistemul nervos central
Sistemul nervos poate fi comparat cu o reţea complexă de nervi şi celule ce transportă informaţii de la nivelul creierului la nivelul măduvei spinării, dar şi de la nivelul diverselor părţi ale corpului către creier.
Acesta este alcătuit atât din sistemul nervos central ce cuprinde encefalul şi măduva spinării, cât şi din sistemul nervos periferic ce curpinde fibre nervoase somatice eferente (motorii) şi aferente (senzitive şi senzoriale), dar şi fibre nervoase vegetative eferente sau aferente (simpatice sau parasimpatice).
Sistemul nervos central este împărţit îndouă compomente majore reprezentate de encefal şi măduva spinării.
Encefalul este localizat în interiorul cutiei craniene şi este alcătuit din patru componente principale reprezentate de:
– Trunchiul cerebral;
– Cerebelul;
– Emisferele cerebrale;
– Diencefalul.
Ȋn structura encefalului intră neuroni dar şi celule de susţinere ce alcătuiesc glia.
La nivelul encefalului se pot identifica două tipuri de substanţe: substanţa cenuşie ce recepţionează şi reţine informaţii şi substanţa albă ce transportă impulsurile nervoase către/ şi de la nivelul substanţei cenuşii.
Emisferele cerebrale sunt în număr de două, fiecare emisferă controlând activitate de la nivelul părţii controlaterale a corpului.
Ele sunt împărţite la rândul lor înşase formațiuni şi anume:
– Lobul frontal;
– Lobul parietal;
– Lobul temporal;
– Lobul occipital;
– Insula Reil;
– Sistemul limbic.
Cerebelul este localizat posterior şi inferior faţă de emisferele cerebrale şi este alcătuit din două emisfere cerebeloase şi vermis.
Trunchiul cerebral prezintă trei porţiuni: bulb, punte şi mezencefal (acesta este alcătuit din pedunculii cerebrali şi coliculii cvadrigemeni).
Diencefalul este compus din mai multe componente şi anume:
– Talamus;
– Hipotalamus;
– Epitalamus;
– Subtalamus;
– Metatalamus.
Talamusul este localizat la nivelul ventriculului III, pe peretele lateral al acestuia şi are forma unui ovoid.
Epitalamusul este alcătuit din comisura albă posterioară, glanda epifiză şi trigonul habenular;
Măduva spinării este localizată la nivelul canalului vertebral realizat prin suprapunerea vertebrelor ce intră în alcătuirea coloanei vertebrale. Măduva spinării este învelită la exterior de meningele spinal reprezentat de cele trei foiţe ale acestuia: dura mater, arahnoida şi pia mater. Măduva spinării are forma unui cilindru turtit si este alcătuită atât din substanţă albă, cât şi din substanţă cenuşie. Substanţa cenuşie este substanţa centrilor şi releelor, substanţa albă este locul unde prelungirile celulare se aglomerează în fascicule şi alcătuiesc aparatul de transmisie.
Măduva spinării prezintă două funcţii principale şi anume:
– funcţia de centru reflex şi
– funcţia de conducere.
Măduva este parcursă de două șanțuri secundare colaterale. Din fiecare șant colateral pleacă un ansamblu de filete nervoase care se grupează în rădăcini. De fiecare parte a măduvei i-au naștere 31 rădăcini posterioare și 31 rădăcini anterioare. Fiecare rădăcina posterioară se unește cu rădăcina anterioară de la același nivel pentru a forma un nerv rahidian. Informațiile senzitive ajung la măduvă prin rădăcinile posterioare ale nervilor.
Lichidul cefalorahidian (LCR) este un lichid transparent și incolor, care se găsește în spațiile ventriculare ale creierului, precum și în spațiul subarahnoidian. (spațiul situat între membrana arahnoidă și pia mater), practic „scăldând” creierul și măduva spinării. Compoziția LCR este: 99% apă, 115 mEq/L NaCl, 0,5 g/L glucoză și 0,4 g/L proteine (albumine, etc). Compoziția sa reflectă starea fiziopatologică a creierului (inflamație, infecție, etc).
Funcții:
LCR are rol de protecție a țesutului nervos împotriva traumatismelor, prin amortizarea unor șocuri la care este supusă cutia craniană.
Sistemul nervos central este format din măduva spinării şi encefal.
Encefalul are un rol major ȋn controlul diferitelor funcţii ale organismului precum motilitatea, sensibilitatea, gândirea, memoria, vorbirea.
Măduva spinării este conectată cu encefalul prin intermediul trunchiului cerebral şi prezintă două funcţii principale şi anume: funcţia de centru reflex şi funcţia de conducere.
Encefalul este ȋmpărţit ȋn mai multe regiuni, fiecare având o funcţie specifică (de exemplu, lobul frontal deţine funcţiile cognitive şi memoria).
Deşi encefalul şi măduva spinării au o simbioză pentru a controla diverse funcţii ale organismului, anumite reflexe pot avea loc doar prin intermediul măduvei spinării, fără implicarea nici unei structuri aparţinând encefalului.
Orice leziune de la nivelul măduvei spinării sau de la nivelul encefalului poate determina afectarea unor functii variate controlate de acestea.
Epilepsia este o afecţiune cronică, caracterizată prin recurenţa crizelor.
Boala epileptică a fost clasificată în sindroame epileptice și alte tipuri de epilepsie . Astfel se diferențiază:
1.sindroame epileptice idiopatice (primare):
– convulsiile neonatale benigne
– epilepsia mioclonica juvenilă
– epilepsia benignă a copiilor cu vârfuri centrotemporala
– sindroame epileptice simptomatice (secundare):
– sindromul Lennox-Gastaut
– sindromul lobului temporal medial
– starea de rău epileptic (status epilepticus)
– epilepsia reflexă (majoră, minoră, sau localizată)
– epilepsia morfeica
– epilepsia catamenială
2.Convulsiile neonatale benigne
Convulsiile neonatale benigne sunt afecțiuni generalizate, idiopatice sesizate la nou născuții normali, între ziua a 2-a și a 6-a de viață. Crizele sunt de obicei tonice sau se caracterizează ca scurte perioade de apnee și se remit spontan în zile sau săptămâni; copii nu vor prezenta sechele.
3.Epilepsia mioclonica juvenilă
Epilepsia mioclonica juvenilă este o afecțiune ce prezintă crize generalizate, de cauză necunoscută, apare în primii ani ai adolescenței și se caracterizează prin paroxisme mioclonice bilaterale, ce pot fi unice sau repetitive. Atacurile mioclonice sunt cel mai frecvent dimineață, după trezire și pot fi provocate de privarea de somn. Conștienta este păstrată dacă miocloniile nu sunt foarte severe. Mulți pacienți au și atacuri tonico-clonice.
4.Epilepsia benignă a copiilor cu vârfuri centrotemporale
Este o epilepsie focală, idiopatică ce apare la copii de altfel normali, cu vârste între 3 și 13 ani și poate ajunge la 25% din epilepsiile din copilărie. Pacienții au crize scurte, parțiale-simple, caracterizate prin simptome motorii și senzitive pe jumătate de față, ce se pot răspândi la membre sau se pot generaliza.
Atacurile sunt ușor de tratat cu anticonvulsivante și se remit aproape întotdeauna până la 15 ani. Boala se transmite autozomal dominant, cu penetranță variabila, iar mulți membri ai familiei aparent neafectați au anomalii EEG.
5. Sindromul Lennox-Gastaut se asociază cu suferință a SNC sau disfuncții din diferite cauze, ce cuprind anomalii de dezvoltare, hipoxie/ischemie perinatală, traumatisme, infecții și alte leziuni dobândite. Natura multifactorială a acestui sindrom sugerează că este un răspuns nespecific al creierului la lezare neuronală difuză. Din nefericire, mulți pacienți au un prognostic rezervat, din cauza bolii SNC subiacente și a consecințelor psihosociale și fizice ale epilepsiei severe, insuficient controlate.
Sindromul Lennox-Gastaut este present la copii cu vârste între 1 și 8 ani și este definit de următoarele caracteristici:
-crize de multe tipuri (ce cuprind de obicei crize generalizate tonico-clonice, atone și absente (atipice);
-EEG ce arată descărcări lente (< 3Hz) vârf-undă și o varietate de alte anomalii
– alterarea funcțiilor cognitive la majoritatea pacienților, dar nu în toate cazurile.
Un sindrom similar la sugari, ce evoluează frecvent în sindrom Lennox- Gastaut, este sindromul West caracterizat prin spasme infantile, alte elemente de disfuncție cerebrală și un model EEG caracteristic.
6. Sindromul epilepsiei lobului temporal medial
Epilepsia lobului temporal medial (ELTM) este cel mai frecvent sindrom asociat cu crize parțiale complexe și este un exemplu de epilepsie parțială simptomatică. Rezonanța magnetică de înaltă rezoluție poate detecta scleroză caracteristică în hipocamp, ce pare ca un element esențial fiziologic al ELTM pentru mulți pacienți; recunoașterea acestui sindrom este foarte importantă, deoarece tinde să fie refractar la tratamentul cu medicamente anticonvulsivante, dar răspunde foarte bine la intervenție chirurgicală. Progrese majore în înțelegerea mecanismelor de bază s-au făcut prin studierea modelelor experimentale ale ELTM.
Sunt cunoscute multe cazuri de epilepsie şi frecvenţa lor variază cu vârsta. La nou născut cauzele cele mai frecvente sunt traumatismele obstreticale, malformaţiile congenitale, tulburări metabolice şi injecţiile. La adulţi şi vârstnici cauzele cele mai frecvente sunt boala aminomusculară, TCC, şi tulburarea aminală degenerativă, tumoricerebrale, droguri şi intoxicaţii. La cel puţin jumătate dintre pacienţi nu se găseşte vreo cauză după investigaţii detaliate; în asemenea cazuri factorii genetici par a avea o semnificaţie mai mare decât cei cu patologie demonstrabilă.
Leziunile cerebrale organice în evoluție sechelară în contextul unui prag epileptogen scăzut poate să provoace epilepsie. În cazul leziunii acute prin suferință neuronală cu tulburări dismetabolice, ischemie, edem, se poate determina hiperexcitabilitatea neuronală, iar în cazul leziunii sechelare cicatricea glio-meningo-vasculară are în imediată apropiere neuroni dezaferentați, cu potențial de dezinhibiție. Populațiile neuronale modificate funcțional de leziunea organică constituie focarul epileptogen.
Cauzele în epilepsia secundară se grupează în cauze perinatale și postnatale.
Cauze perinatale:
– infecții materne
– iradieri abdominale cu raze Roetngen
– traumatisme abdominale
– tentative de avort
– disgravidii severe
– icter nuclear prin compatibilitate Rh
Factori natali:
– traumatisme obstericale,
– aplicări de forceps cu vacum extractor cu determinarea de contuzii cerebrale sau hematoame
– expulzii brutale
– anoxii cerebrale fetale
Cauze postnatale:
– factori postnatali imediați: malformații cerebrale, hemoragii subarahnoidiene neonatale primare, infecții neonatale meningo-encefalice.
– traumastismele cranio-cerebrale sunt găsite în 15-40% din toate epilepsiile secundare. În funcție de intervalul impact cranio-cerebral-epilepsie crizele post-traumatice pot fi împărțite în crize precoce, care survin în prima lună, și crize tardive. Crizele posttraumatice precoce pot să apară la câteva ore, zile sau săptămâni de la impact, ele constituind dovadă clinică a unui proces distructiv cerebral sau a unei complicații că hematomul sau abcesul, care pot fi înlăturate chirurgical cu efecte pozitive asupra crizelor. Crizele posttraumatice tardive sunt determinate de cicatrice epileptogene.
– tumorile cerebrale sunt întâlnite în 7-12% din cazurile de epilepsie simptomatică, cifra care crește la 40% din cazurile de epilepsie cu debut peste vârsta de 40 de ani. Tumorile cerebrale suprazenzoriale prezintă în tabloul lor în 40-50% crize epileptice. Localizarea fronto-parietală a tumorilor cuprinde cel mai adesea și epilepsie.
– bolile cerebro-vasculare intervin în procent de 27% din epilepsiile secundare.Apar în toate tipurile de boală vasculară, în perioada acută și în faza sechelara.
– bolile infecțioase se întâlnesc în etiologia epilepsiei secundare atât în cazul abcesului și a tuberculomului cerebral, cât și în meningite și encefalite primitive virale sau în encefalite secundare, în infecții virale lente și în parazitoze cerebrale (echinocoza, trichinoză, cisticerocoza).
– disembrioziile (facomatoză) boli genetice cu caracter ușor evolutiv.
Produc frecvent epilepsie:
– scleroză tuberculoasă (boala Bourneville)
– angiomatoză encefalotrigerminala (boala Sturge-Weber Krabbe)
– neurofibromatoză Recklinghausen
Diagnosticul manifestarilor psihice critice in epilepsie se sprijina pe mai multe categorii de fapte ce necesita o analiza amanuntita.
1. EEG –localizeaza focarul epileptic,ajuta la clarificarea tipului de criza.
2. RMN,CT identifica eventualele leziuni cerebrale.
Prognosticul si evolutia epilepsiilor depinde de vârstă, etiologie, evoluţia terapeutică şi alţi factori care pot fi importanţi de la caz la caz. Mortalitatea este relativ ridicată la bolnavii epileptici, în particular în epilepsiile cu debut în primul an, epilepsiile simptomatice şi SI.
Cauzele de deces sunt:
direct legate de crize sau SE (10%),
accidente în timpul crizelor (5%),
suicid (7-22%),
moarte subită (> 10%).
Dintre sindroamele epileptice pediatrice 70% pot fi vindecate cu o terapie corectă.
Tratamentul epilepsiilor este complex şi constă în:
1.tratament farmacologic;
2.asistenţă psihosocială;
3.tratament chirurgical în formele farmacorezistente.
Terapia antiepileptică este simptomatică, scopul fiind obţinerea unui control cât mai bun posibil al crizelor. Tratamentul trebuie aplicat cât mai precoce, deoarece repetarea crizelor contribuie la consolidarea mecanismelor patogenice ce intervin în declanşarea lor. Evaluarea rezultatelor se va baza în primul rând pe criteriul clinic şi ulterior pe cel EEG.
1.Terapia farmacologică. Există un număr însemnat de MAE între care unele sunt de ordinul I adică administrate singure ca monomedicaţie pot controla crizele, iar altele sunt de ordinul II ce nu vor reuşi să controleze crizele decât în asociere cu un alt MAE .
Tratamentul trebuie întotdeauna iniţiat prin monoterapie. Dozele unui singur MAE sunt crescute progresiv până la atingerea unei concentraţii serice terapeutice fără efecte adverse semnificative. În cazul crizelor care nu răspund la tratament, trebuie folosită monoterapia cu un alt MAE care va fi introdus progresiv ca şi primul. După obţinerea controlului crizelor primul MAE se va suspenda lent aşa cum a fost introdus. Politerapia va fi instituită numai în cazul eşecului monoterapiei cu MAE de primă linie.
După câţiva ani de tratament (2-4 ani) cu un bun control al crizelor se pune problema diminuării terapiei , ce se va face foarte lent începând cu 1/4 sau 1/5 din doza zilnică, la 4-6 luni interval, asociat cu un control EEG
Crizele epileptice nu sunt în general, periculoase și se opresc după câteva minute. Dacă o criza generalizată durează mai mult de 15 minute, e posibil ca circulația sângelui în creier să fie afectată.
Status epilepticus este un tip de epilepsie în care criza durează mai mult de 20 de minute, iar pacientul prezintă crize convulsive intermitente , consecutive fără să-și recapete cunoștința între timp.
Epilepsia afectează diferit fiecare pacient în parte. Cei mai mulți pacienți au o viată normală, activă, putând alege fără restricții orice activitate pentru timpul liber. Activitătile sportive și gradul lor de risc: alpinismul sau simplele plimbări pe munte sunt riscante, boxul, artele marțiale, luptele care implică un risc crescut de lovituri în zona craniană nu sunt recomandate persoanelor epileptice; înotul în mare, râuri sau lacuri este periculos din cauza curenților, valurilor, variațiilor de temperatură a apei;
Esențiale pentru primul ajutor în cazul crizelor tonico-clonice generalizate sunt următoarele:
-păstrarea calmului și liniștirea celor din jur;
-limitarea miscărilor pacientului în timpul crizei;
-inlăturarea obiectelor rigide din jurul pacientului pentru a nu suferi un traumatism;
-așezarea sub capul pacientului unui obiect textil pentru protecția de lovituri;
-întoarcerea pacientului pe o parte pentru menținerea căilor respiratorii libere.
-supravegherea pacientul pană la epuizarea crizei și revenirea stării de conștientă.